东方富海博士后创新实践基地秉承研究发现价值、研究引领投资的理念,对投资实务进行前瞻性研究。我们特别设立了“富海洞察”专栏,用于发布基地系列研究报告,供交流探讨。本文是靶向药物开发的相关研究,为基地出品的第六篇报告。
在过去的几十年中,小分子靶向药的开发策略主要是通过与靶基因目标蛋白的相互作用(抑制或者激活)来发挥治疗作用,也就是靶蛋白的抑制剂或者激活剂。理想情况下,小分子靶向药应该对正常组织没有毒性。实际上,由于选择性和特异性问题,小分子靶向药仍然具有不良的毒性和副作用:药物本身对目标蛋白的特异性较低,对其他蛋白质具有脱靶活性,或者目标蛋白不是组织特异性的,在其他正常细胞中具有生理功能。在抗肿瘤小分子靶向药的实际临床应用中,还会遇到一个非常棘手的问题,就是肿瘤的耐药性问题。
最常见的肿瘤耐药机制就是突变。通过对某些位点的基因突变,目标蛋白会丧失对药物的敏感性,产生耐药。另一种耐药机制是,肿瘤通过对目标蛋白的代偿性高表达、或者激活其他通路来逃避药物的作用,从而产生耐药。以上的局限性,限制了小分子靶向药在各种疾病,尤其是抗肿瘤领域中的应用。为了克服抑制剂/激活剂型的小分子靶向药的局限性,基于蛋白降解的小分子药物逐渐被人们重视。
1PROTAC的发展背景
蛋白水解靶向嵌合体(PROTACs)技术是近年来出现的一种新的蛋白质降解策略。它使用双功能小分子通过泛素-蛋白酶体系统诱导靶蛋白的泛素化和降解。PROTACs不仅可以作为癌症、免疫紊乱、病毒感染和神经退行性疾病等疾病的潜在临床治疗方法,而且可以以催化、可逆和快速的方式为生物学研究提供独特的工具。
PROTAC(蛋白降解靶向联合体)最早是在2001年由Crews等人提出的。PROTAC技术在二十多年来的发展中,已逐渐发展为当今药物研究中最为炙手可热的技术,受到科研单位、药企和投资商的广泛关注。
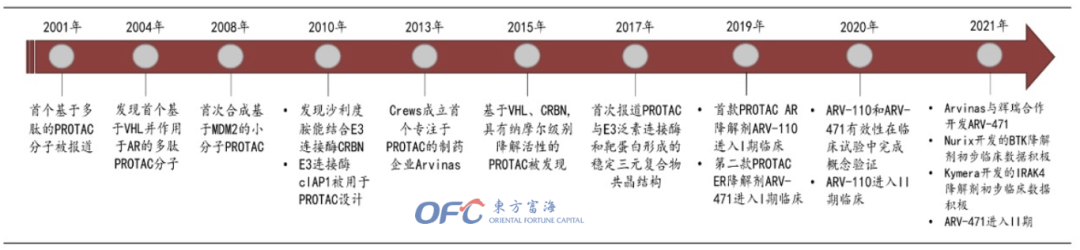
▲PROTAC的发展历史
PROTAC是一种特异性双功能分子,分子的一端连接结合靶蛋白的配体,一端连接E3连接酶的配体,中间通过合适的Linker相连。
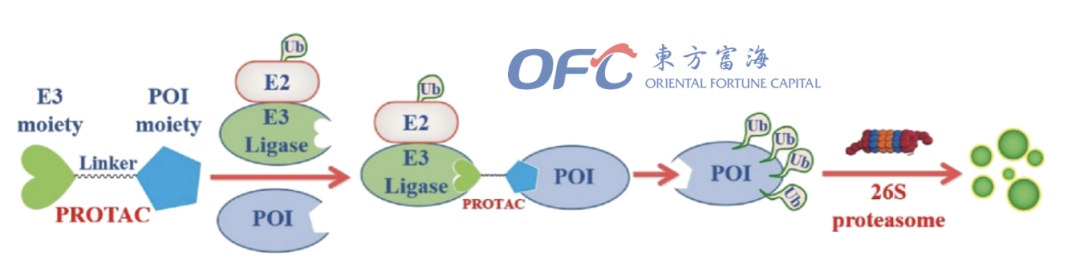
▲PROTAC介导蛋白降解的机制
PROTAC降解靶蛋白是通过劫持泛素蛋白酶体系统(UPS)实现的。其大致过程如下,PROTAC分子结合靶蛋白(POI)和E3连接酶,形成三元复合物,给靶蛋白打上泛素化的标签,泛素化的蛋白被细胞内的蛋白酶体识别并降解。
与其他药物的浓度-活性曲线不一样的是,PROTAC的药物浓度与降解活性呈现一个特殊的“钟”型曲线。PROTAC在较大浓度的时候优先形成的是目标蛋白-PROTAC和E3-PROTAC两种二元复合物,影响三元复合物的形成,所以PROTAC的降解活性并不是严格的浓度依赖性,在较大浓度时会出现降解活性下降的反常现象。

▲PROTAC的钟型曲线模型
PROTAC技术是目前最为有效的,蛋白质层面的基因降解技术,是基于核酸的基因敲除/敲降技术的有效补充。
2PROTAC的优点
1靶点更广泛:
传统的小分子和抗体等都是通过“占据驱动”的作用模式抑制靶蛋白的功能发挥治疗疾病的作用,这种作用模式需要抑制剂或单抗具备较高的浓度才能够占据靶点的活性位点,阻断下游信号通路的转导。而PROTAC是“事件驱动”,不是影响蛋白的功能,而是介导致病靶蛋白被降解。只要PROTAC介导三元复合物的形成并给靶蛋白打上泛素化的标签,理论上是可以循环反复使用的,因此催化剂量即可发挥作用。而且PROTAC对于没有活性位点的蛋白,如支架蛋白等,只要能够产生结合作用就可以诱导相关蛋白被降解,可以大大提高靶点的范围。
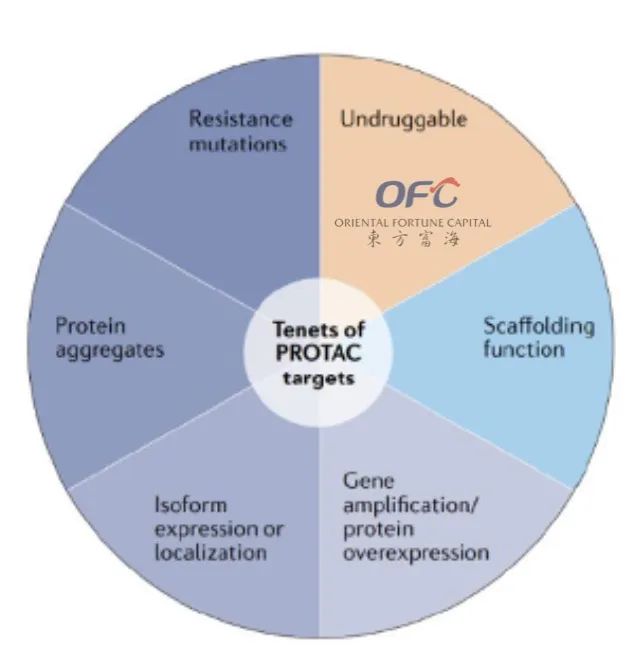
▲适合PROTAC的蛋白靶标类型
适合PROTAC的靶标主要满足四个特征:
第一个是“不可成药”;第二个是蛋白表达偏离自然状态(过表达、突变、异常聚集、折叠错误等);第三个是支架蛋白;第四个是已经对现有的疗法和药物出现耐药性。
据不完全统计,目前已经有超过100种蛋白被成功降解。这些靶点包括激酶类,如RIPK2、BCR-ABL、EGFR、HER2、c-Met、TBK1、CDK2/4/6/9、ALK、Akt、CK2、ERK1/2、FLT3、PI3K、BTK、Fak等;BET蛋白,如BRD2/4/6/9;核受体,如AR、ER等;其他蛋白,如MetAp-2、Bcl-xL、Sirt2、HDAC6、Pirin、SMAD3、ARNT、PCAF/GCN5、Tau、FRS2等。这其中就包括“不可成药靶点”,如转录因子调节蛋白pirin、表观遗传相关蛋白PCAF/GCN5等。

▲人类激酶谱(标红为可被PROTAC降解)
根据人类激酶图中标记的激酶,我们可以发现,除了CK1和CAMK类的激酶,其余的激酶大类都有被PROTAC降解的记录,尤其是第一大类激酶RTKs和第二大类CMGCs。
2选择性更高、安全性更高:
与传统小分子抑制剂相比,PROTAC在某些靶点上可实现小分子难以实现的选择性。众所周知,小分子抑制剂往往不止一个抑制靶点,比如激酶抑制剂常常都以2,4-二氨基嘧啶为骨架,EGFR、ALK、CDK、Jak等激酶的抑制剂都有用到其作为母核。我们通常说这类药物分子比较“脏”,也就是靶点选择性比较差,经常对其他很多激酶都有很强的抑制活性,开发过程中off target的脱靶效应常常成为毒副作用的主要来源,影响新药开发的成功率。
3可以克服耐药性:
小分子抑制剂或拮抗剂在临床用药过程中,不可避免地都会发生获得性耐药。比如EGFR-T790M和C797S耐药等。虽然可以通过开发新一代的抑制剂,如第三代和第四代EGFR来解决耐药问题,但是随着新一代药物的使用,新的耐药也会随之出现。PROTAC技术在克服耐药方面已经显现出了一定的优势,全球有数十个研究小组在尝试关于EGFR-PROTACs的研究,旨在通过蛋白降解途径克服耐药突变或找寻突破抑制剂的蛋白降解疗法。从这些研究成果中,我们可以看到,第一、二、三、四代抑制剂作为结合靶蛋白EGFR的配体,都有被应用到PROTAC的设计中。
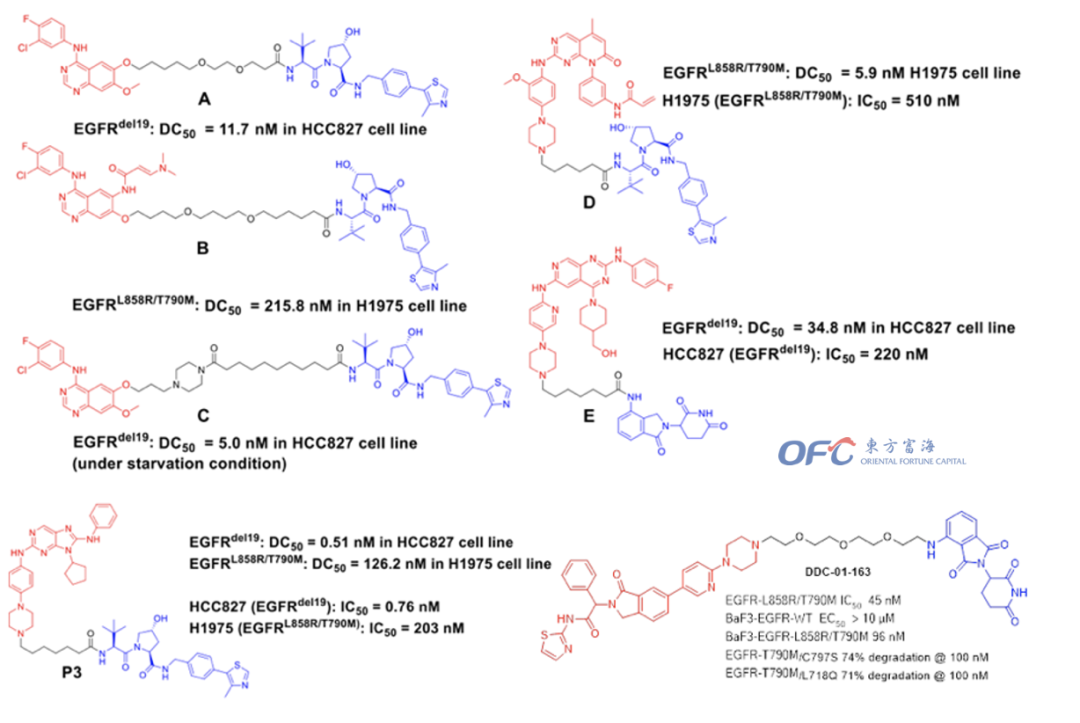
▲若干针对EGFR耐药突变的PROTAC设计
3PROTAC面临的挑战
自2001年首次被人设计出来后,PROTAC技术从概念进入实际应用阶段。尽管PROTAC领域在过去的20年里得到了快速发展,但仍有许多挑战需要解决。这些挑战主要来自以下几个方面,即PROTAC的靶点选择、分子设计、成药性的优化,以及生物活性的综合评价:
(1)PROTAC药物分子量一般在700~1200之间。较大的分子量意味着较差的透膜能力与较差的生物利用度,这极大地限制了PROTAC的口服化和体内活性。
(2)与传统小分子药物不同,目前尚无有效的高通量筛选技术用于快速、大量地评估PROTAC降解POI的能力,只能通过细胞活性筛选或免疫印迹实验等方法实现,这大大降低了开发PROTAC的速度与成功率。
(3)由于PROTAC发挥作用需要形成【目标蛋白-PROTAC-E3酶】三元复合物,很难得到准确的复合物晶体结构,所以目前对PROTAC结构改造缺少指导方向,更多地依靠经验摸索。
(4)发现与E3酶受体蛋白特异性结合的底物具有偶然性。目前报道的PROTAC主要靶向CRBN与VHL,尽管CRBN与VHL是否会发生突变以及突变后是否对PROTAC有影响还是未知,但了解新的E3酶并开发相应的PROTAC具有重要意义,也面临巨大挑战。
4.PROTAC的设计
1蛋白配体:
目标蛋白的配体一般选择上市或者文献报道的具有一定活性的抑制剂,按照抑制剂能否形成共价键,可分为可逆抑制剂、共价不可逆抑制剂和共价可逆抑制剂;根据结合口袋的不同,又可分为ATP竞争性抑制剂和变构抑制剂。为了获得具有知识产权的PROTAC,常常先对抑制剂做一定的结构衍生,使用优化后的抑制剂作为目标蛋白的配体。比如,Crews组通过刚性哌嗪接头将Vemurafenib(威罗菲尼)与VHL偶联,开发了基于VHL的PROTAC。

▲SJF-0628分子结构和基本性能
2E3连接酶及配体:
目前文献报道的应用到PROTAC中的E3连接酶主要有CRBN、VHL、cIAP和MDM2。效果较好、使用频次最高的E3连接酶主要是CRBN和VHL两种。
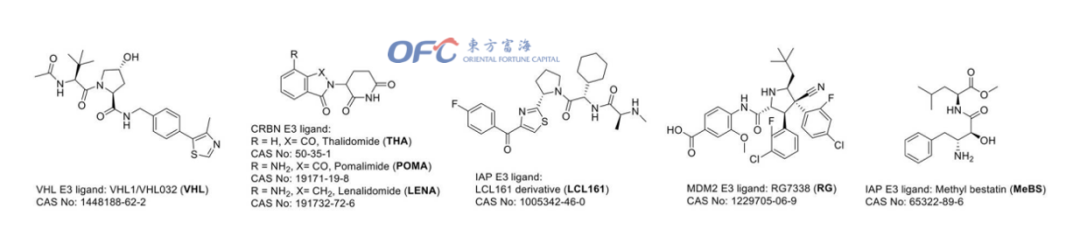
▲常见E3连接酶的常用配体结构
CRBN的配体主要是沙利度胺、泊马度胺、来那度胺及其类似物。VHL的配体主要是VHL-1和一些高VHL结合力的拟肽。IAP常常在肿瘤组织中高表达,并促进癌细胞的生存,所以也是PROTAC开发的重点E3酶之一。
3 Linker:
根据Linker构成的不同,可分为烷基链和PEG链,也有文献使用刚性更强的炔基双哌啶环、含氮原子的螺环或桥环等作为Linker,以限制PROTAC分子的柔性和自由度。已有的文献报道表明,Linker的长短也会影响PROTAC的降解活性,常用的Linker长度一般在4~15个碳原子(或杂原子)。根据作用靶点的不同,Linker的长短对降解活性的影响也不同。此外,Click chemistry(点击化学)由于反应条件较温和、效率较高,常常被应用到PROTAC分子的Linker当中,用于连接两端的分子。
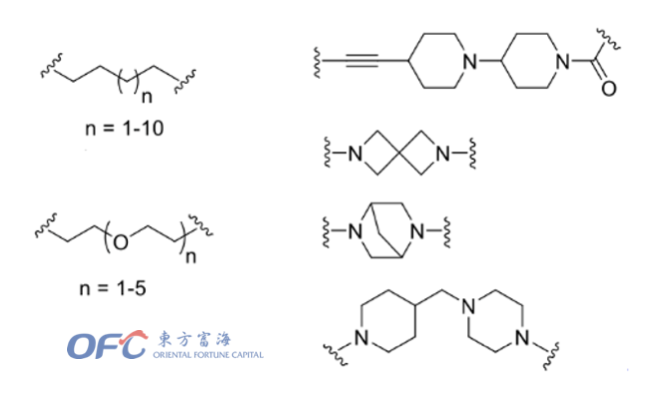
▲常见的PROTAC linker
4连接位点的选择与构成:
PROTAC结构上是通过Linker连接两个配体,除了Linker的构成和长短会影响降解活性外,Linker连接的位点也会影响降解活性,甚至影响选择性。
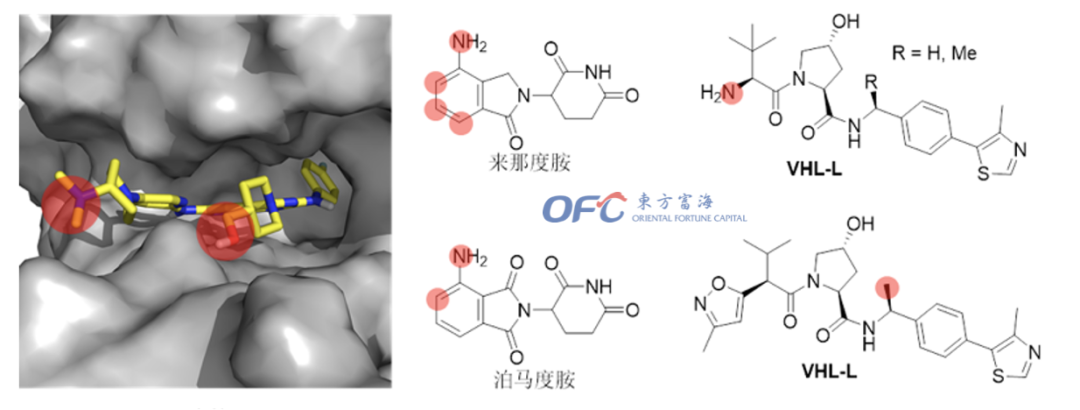
▲PROTAC连接位点的选择
目标蛋白的配体和E3连接酶配体的连接位点一般是在配体暴露在溶剂的区域,通过酰胺键、碳原子或杂原子(如O、N等)等连接,通过缩合反应或亲核取代反应等来实现连接。
5国内布局PROTAC的企业
国内有十余家企业布局PROTAC赛道,其中也不乏一些大型药企和上市公司。
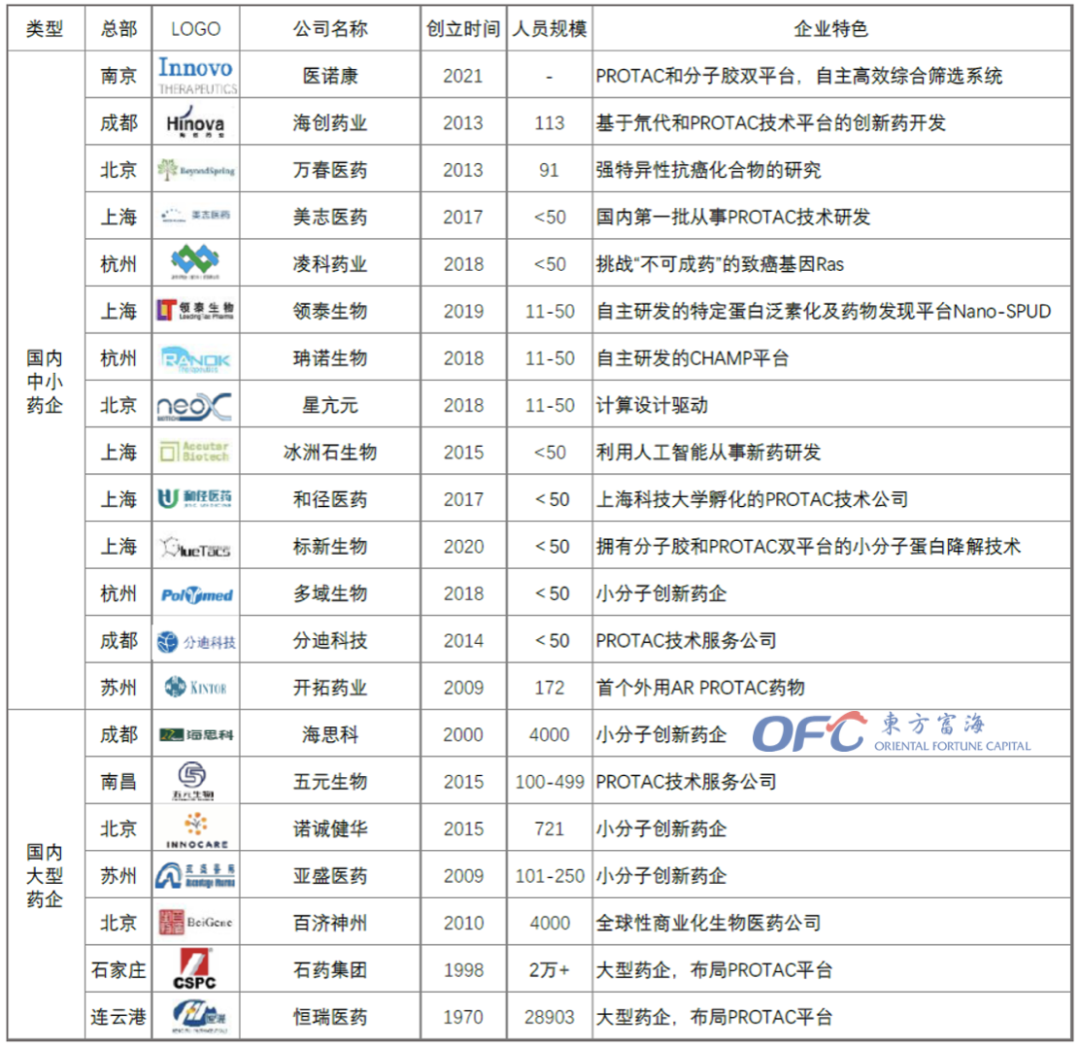
▲国内布局PROTAC的企业
从企业成立时间不难发现,绝大多数布局PROTAC的企业成立于2017年之后,尤其是集中于2017-2019年,这也是PROTAC的基础研究成果井喷式发展的时间段。2019年,全球第一款PROTAC药物ARV-110进入临床I期。
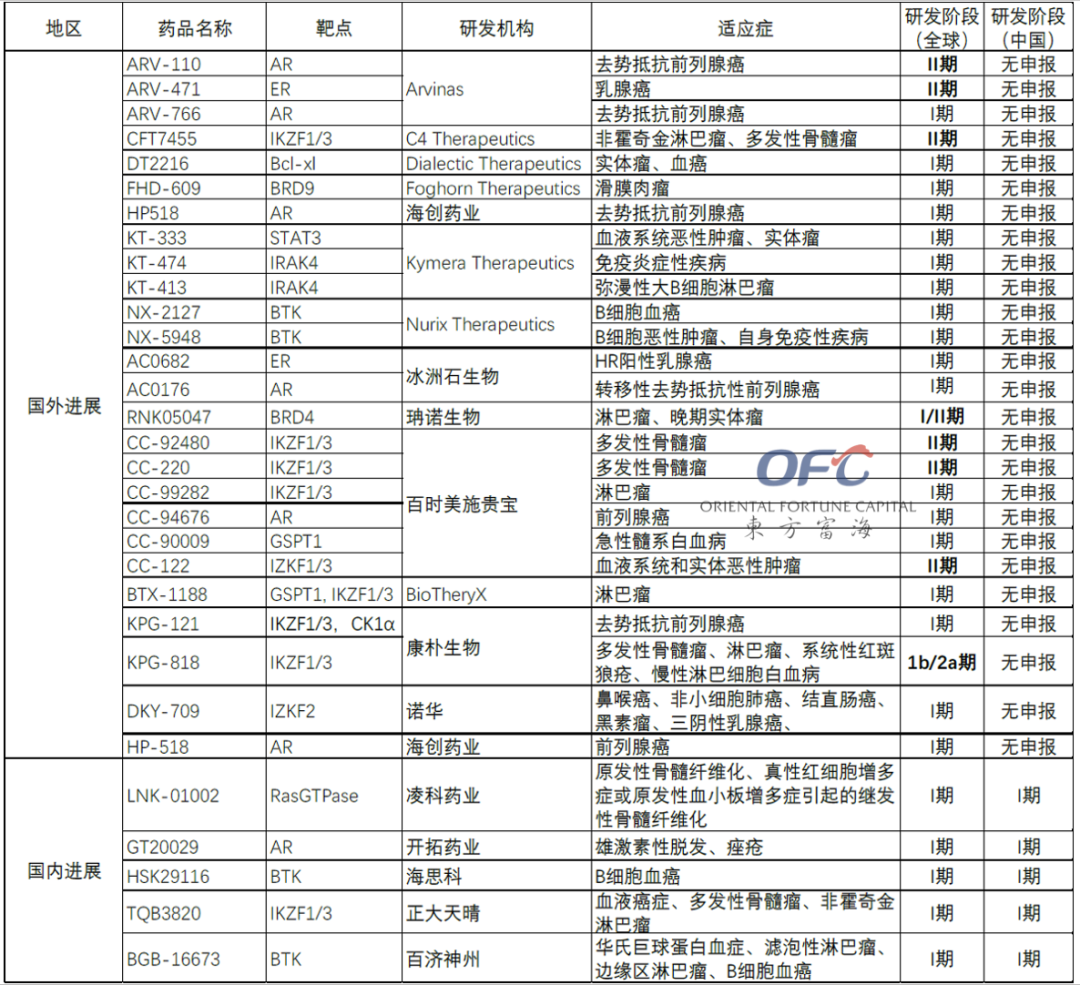
▲PROTAC管线临床进展
截至2022年5月,全球进入临床阶段的PROTAC药物共有31个左右。从药物靶点上看,靶点的集中度很高,并且都是临床上已经过大量验证的靶点,策略十分稳健。
目前进展最快的PROTAC药物是由Arvinas开发的ARV-110和ARV-471,其靶点选择和分子设计堪称PROTAC药物的典范。Arvinas作为蛋白降解领域的先驱,是PROTAC管线推进最快的公司,由PROTAC鼻祖Craig Crews教授创办。2021年4月,Arvinas终于公开了两个明星管线的分子结构,为PROTAC赛道的发展指明了方向。
ARV-110
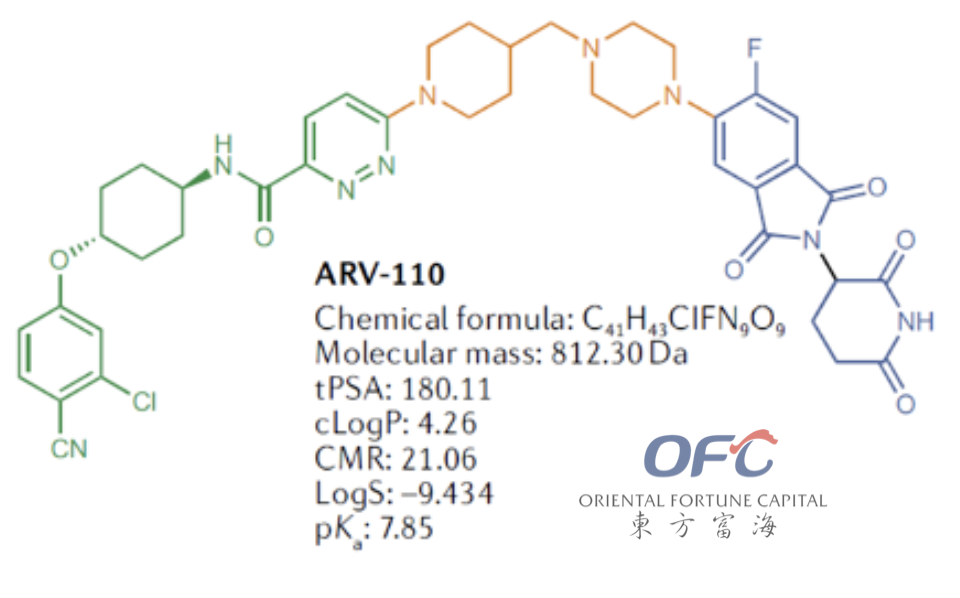
▲ARV-110分子结构
ARV-110是Arvinas在蛋白降解靶向嵌合体领域的全球首个进入临床试验的口服生物可利用的PRAOTC小分子药物,选择性靶向降解雄性激素受体(AR)。在2019年5月ARV-110获得FDA快速通道批准,主要用于治疗患有转移性趋势抵抗性前列腺癌(mCRPC)。
在发达的欧美地区,mCRPC是男性中第二高发的恶性肿瘤,也一直是前列腺癌领域的治疗难点和临床研究的热点。目前主要针对雄性激素受体靶点(AR)的一线治疗药物有阿比特龙和恩杂鲁胺。
在临床前研究中,ARV-110已显示出有希望的活性作为AR的靶向降解剂。在对恩杂鲁胺敏感的模型中,ARV-110表现出与恩杂鲁胺类似的前列腺特异性抗原(PSA)减少,且用药剂量更低。在恩杂鲁胺抗性模型中,ARV-110能明显抑制肿瘤生长。
2022年2月,Arvinas 发布的临床1期剂量爬坡和临床2期中期数据显示,420 mg的剂量确定安全,正在进行的2期临床中沿用该剂量。ARV-110能在体内作用于特定靶点,并且初步显示出治疗肿瘤的效果:在携带 AR T878X/H875Y 突变的肿瘤患者中,ARV-110 使46%患者的前列腺特异性抗原(PSA)水平降低≥50%(PSA50);在符合RECIST标准、可评估的7名患者中,6名患者的肿瘤减小,2名患者达到部分缓解。Arvinas计划在2022 年底开展关键性临床 3 期试验,在携带 AR T878X/H875Y突变的肿瘤患者中进一步评估ARV-110 的治疗效果。
ARV-471
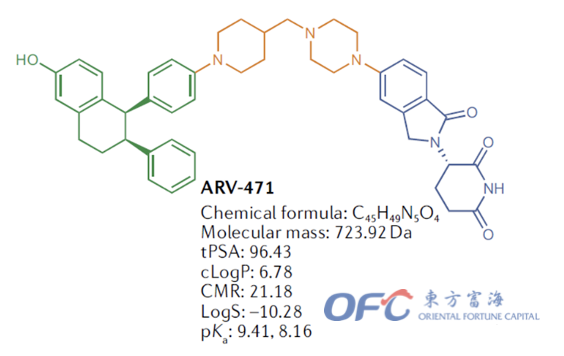
▲ARV-471分子结构
乳腺癌作为一种发生在乳腺细胞中的恶性肿瘤,是近年来致死率位居女性恶性肿瘤首位的癌症。根据2020年全球肿瘤流行病统计(GLOBOCAN2020),在女性患者中乳腺癌发病率位居首位,为24.5%。ARV-471是Arvinas和辉瑞共同开发的雌激素(ER)降解剂,用于治疗ER+/HER2-乳腺癌。
根据2021年12月10日Arvinas发布的临床 1 期结果,在患者入组前都接受过多种治疗的前提下,ARV-471具有良好的安全性,每日口服一次用药,剂量爬坡实验中700 mg没有观察到剂量限制毒性。
相比目前临床中使用的ER降解剂氟维司群(fulvestrant)40~50%的降解率,ARV-471的降解率平均值为 64%,最高值为89%。在可评估的47名入组患者中,19名临床获益(包括部分缓解和病情稳定),获益率为40%,其中3名患者部分缓解。
6未来的趋势
蛋白降解领域明星公司Arvinas的联合创始人之一,耶鲁大学的Craig M. Crews教授和合作伙伴在Nature Reviews Drug Discovery上联合发表深度综述,展望了PROTAC靶向蛋白降解技术未来20年的发展方向。他认为,未来20年PROTAC技术主要有四大发展方向。
1确立最适合蛋白降解的靶点类型:
第一波临床期蛋白降解剂的靶点为已经获得临床验证的成熟靶点,比如Arvinas公司选择的雄激素和雌激素受体。针对这些靶点的成功确立了蛋白降解剂成为一种治疗模式。然而,这一治疗模式真正的潜力在于触达目前治疗模式难于成药或不可成药的靶点。最适靶点的确立将在很大程度上推动PROTAC在临床上的应用。
2扩展可用的E3连接酶:
目前在研的PROTAC大多使用CRBN E3连接酶,少部分招募VHL。
多个因素可能影响连接酶的降解特征,包括连接酶的构象和靶点蛋白之间的互补性;连接酶与靶点蛋白直接构成可导致降解的三元结构的能力;不同连接酶和靶点蛋白的细胞内定位;以及细胞类型特异性连接酶和靶点的表达。
基于连接酶的结构特征,可以判断一些值得用于PROTAC蛋白降解的连接酶,而去年DeepMind和RoseTTAFold在基于氨基酸序列预测蛋白三维结构方面的突破,可能重新定义哪些连接酶可以用于开发蛋白降解疗法。
利用连接酶的组织和细胞特异性,以及在肿瘤中的富集程度和是否对肿瘤必不可少,可能为针对特定领域的蛋白降解疗法提供开发机会。
3扩展肿瘤以外的适应症:
目前在研的管线绝大多数都是以肿瘤为适应性,不过目前PROTAC的适用范围正在向肿瘤以外的领域扩展。
首先就是像炎症和免疫相关的方向。在免疫检查点抑制剂获得成功之后,开发能够激发抗癌免疫反应的小分子药物是药物开发的一个重要领域。PROTAC分子具有以小分子药物的模式激活免疫细胞,模拟PD-1/PD-L1靶向疗法效果,成为潜在“first-in-class”疗法的潜力。最近,靶向MAP4K1的PROTAC分子已经表现出可喜的临床前活性。
其次,在神经生物学和神经退行性疾病方面也有一定的发展空间。PROTAC分子的关键性特征之一是能够降解因为没有活性位点而不被传统小分子抑制剂靶向的蛋白。这一特征让在多种神经退行性疾病中积累的蛋白(比如Tau蛋白,α突触核蛋白,亨廷顿蛋白突变体…)成为潜在靶点。
在抗病毒方面,PROTAC也有自己的一席之地。抗病毒PROTAC分子的可行性最初在降解丙肝病毒(HCV)NS3/4A蛋白酶的研究中得到确立。随着新冠病毒的出现,这一策略也被用于靶向新冠病毒。新冠病毒的主要蛋白酶(Mpro和PLpro)和RNA依赖性RNA聚合酶(RdRP)目前被小分子抑制剂靶向。它们可能成为PROTAC分子的潜在靶点。
4开发创新的PROTAC模式:
创新PROTAC策略,包括基于bioPROTACs和hybrid PROTACs可能进一步扩展PROTAC可以靶向的靶点范围。
bioPROTACs利用多肽、融合蛋白、以及寡核苷酸等作为识别靶点的配体。基于融合蛋白或者多肽的PROTACs已经在研究中成功降解HER2、MYC、KRAS等与癌症相关的靶点。基于寡核苷酸的PROTAC可以用于靶向转录因子和RNA结合蛋白。这些蛋白如果出现突变,会导致多种类型的癌症,以及肥胖症、心血管和神经疾病。
这些bioPROTACs有潜力成为一种新的治疗模式,不过它们的递送和给药模式是需要跨越的障碍。这些疗法可能需要以基因疗法的方式递送,利用精准调控的启动子来控制细胞内的表达水平。基于纳米颗粒的递送技术和基于病毒载体的基因递送系统的发展将在这一治疗模式的进展中起到重要作用。
7总结
目前进展最快的PROTAC药物仍在临床II期,先行者将会承担更多的药理、毒理、生化特性,乃至商业化等诸多方面的问题。
总的来说,国内的PROTAC蛋白降解赛道尚处于起步阶段,作为小分子药物的重要技术路径,参与者仍然较少。无论从玩家数量还是从产业投入上,相对于热门赛道(细胞基因治疗、大分子药物),PROTAC仍然有非常大的发展空间。



